
ML model Interpretation with Molecule Similarity Maps
Posted on * • 5 minutes • 969 words
RDKit’s similarity map functionality (https://jcheminf.biomedcentral.com/articles/10.1186/1758-2946-5-43). Can be used to interpret a machine learning model which is very important for designing new molecules.
Similarity Maps - Intro
from rdkit import Chem
from rdkit.Chem import Draw
from rdkit.Chem.Draw import SimilarityMaps
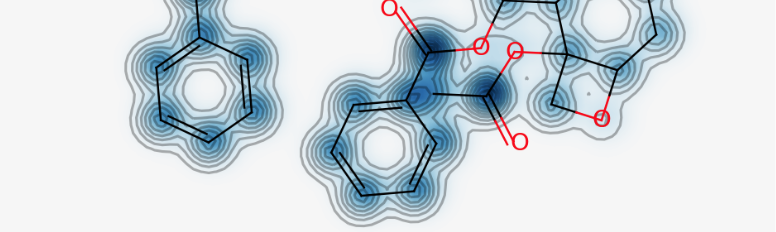
Similarity map functionality using the Morgan fingerprint.
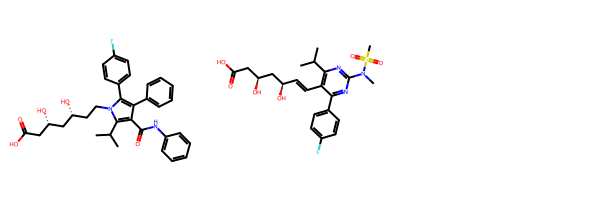
for similarity between atorvastatin (Lipitor) and rosuvastatin (Crestor)
atorvastatin = Chem.MolFromSmiles('O=C(O)C[C@H](O)C[C@H](O)CCn2c(c(c(c2c1ccc(F)cc1)c3ccccc3)C(=O)Nc4ccccc4)C(C)C')
rosuvastatin = Chem.MolFromSmiles('OC(=O)C[C@H](O)C[C@H](O)\C=C\c1c(C(C)C)nc(N(C)S(=O)(=O)C)nc1c2ccc(F)cc2')
Draw.MolsToGridImage((atorvastatin,rosuvastatin))
import io
from PIL import Image
import numpy as np
def show_png(data):
bio = io.BytesIO(data)
img = Image.open(bio)
return img
d = Draw.MolDraw2DCairo(400, 400)
_, maxWeight = SimilarityMaps.GetSimilarityMapForFingerprint(atorvastatin, rosuvastatin,
lambda m, i: SimilarityMaps.GetMorganFingerprint(m, i, radius=2, fpType='bv'),
draw2d=d)
d.FinishDrawing()
show_png(d.GetDrawingText())
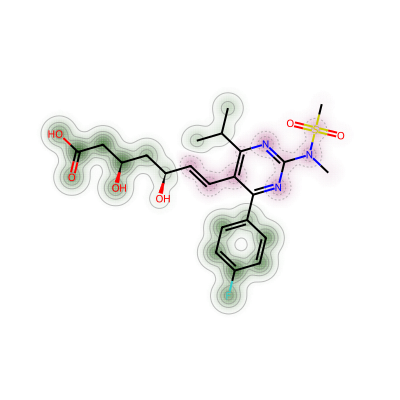
Similarity map functionality using the count-based fingerprints.
d = Draw.MolDraw2DCairo(400, 400)
_, maxWeight = SimilarityMaps.GetSimilarityMapForFingerprint(atorvastatin, rosuvastatin,
lambda m, i: SimilarityMaps.GetMorganFingerprint(m, i, radius=2, fpType='count'),
draw2d=d)
d.FinishDrawing()
show_png(d.GetDrawingText())
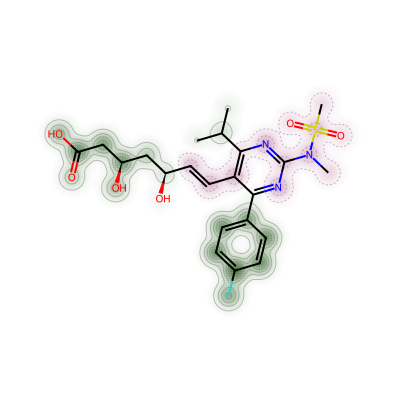
Viewing partial charges
the partial charges calculated with extended Hueckel theory (eHT) using Mulliken analysis
from rdkit.Chem import rdEHTTools
from rdkit.Chem import rdDistGeom
mh = Chem.AddHs(atorvastatin)
rdDistGeom.EmbedMolecule(mh)
_,res = rdEHTTools.RunMol(mh)
static_chgs = res.GetAtomicCharges()[:atorvastatin.GetNumAtoms()]
d = Draw.MolDraw2DCairo(400, 400)
SimilarityMaps.GetSimilarityMapFromWeights(atorvastatin,list(static_chgs),draw2d=d)
d.FinishDrawing()
show_png(d.GetDrawingText())
!!! Warning !!! Distance between atoms 46 and 6 (0.979791 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.996861 A) is suspicious.
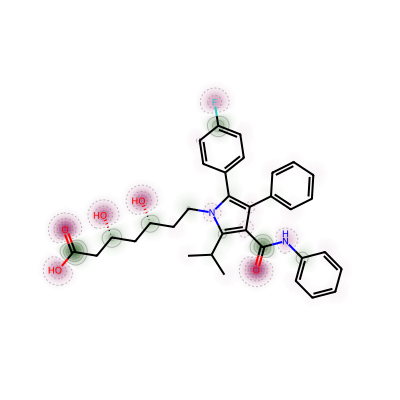
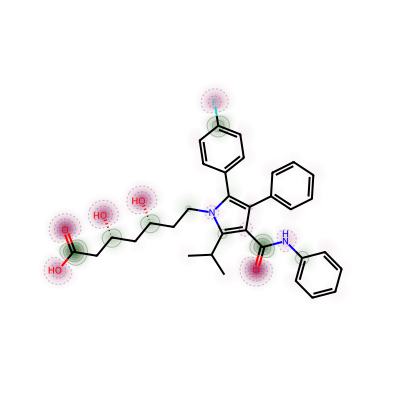
Start by generating 10 diverse conformers, calculating the charges for each, and plotting the average:
mh = Chem.AddHs(atorvastatin)
ps = rdDistGeom.ETKDGv2()
ps.pruneRmsThresh = 0.5
ps.randomSeed = 0xf00d
rdDistGeom.EmbedMultipleConfs(mh,10,ps)
print(f'Found {mh.GetNumConformers()} conformers')
chgs = []
for conf in mh.GetConformers():
_,res = rdEHTTools.RunMol(mh,confId=conf.GetId())
chgs.append(res.GetAtomicCharges()[:atorvastatin.GetNumAtoms()])
chgs = np.array(chgs)
mean_chgs = np.mean(chgs,axis=0)
std_chgs = np.std(chgs,axis=0)
d = Draw.MolDraw2DCairo(400, 400)
SimilarityMaps.GetSimilarityMapFromWeights(atorvastatin,list(mean_chgs),draw2d=d)
d.FinishDrawing()
show_png(d.GetDrawingText())
Found 10 conformers
!!! Warning !!! Distance between atoms 46 and 6 (0.970024 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.981406 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.981542 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.979128 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.990523 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.988965 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.996235 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.971296 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.991920 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.977223 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.984263 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.987732 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.989019 A) is suspicious.
!!! Warning !!! Distance between atoms 46 and 6 (0.970647 A) is suspicious.
!!! Warning !!! Distance between atoms 50 and 9 (0.989967 A) is suspicious.
Random Forest Classifier
%matplotlib inline
import matplotlib.pyplot as plt
import os
from functools import partial
from rdkit import Chem
from rdkit import rdBase
from rdkit import RDPaths
from rdkit.Chem import Draw
from rdkit.Chem import AllChem
from rdkit.Chem import DataStructs
from rdkit.Chem.Draw import SimilarityMaps, IPythonConsole
from IPython.display import SVG
import io
from PIL import Image
import rdkit
import numpy as np
from sklearn.ensemble import RandomForestClassifier
# Load solubility data which is provided by rdkit data.
trainpath = os.path.join('./solubility.train.sdf')
testpath = os.path.join('./solubility.test.sdf')
train_mols = [m for m in Chem.SDMolSupplier(trainpath) if m is not None]
test_mols = [m for m in Chem.SDMolSupplier(testpath) if m is not None]
val_dict = {'(A) low':0,
'(B) medium':1,
'(C) high':2}
def mol2arr(mol, fpfunc):
fp = fpfunc(mol)
arr = np.zeros((0,))
DataStructs.ConvertToNumpyArray(fp, arr)
return arr
# https://rdkit.blogspot.com/2020/01/similarity-maps-with-new-drawing-code.html
def show_png(data):
bio = io.BytesIO(data)
img = Image.open(bio)
return img
fpfunc = partial(SimilarityMaps.GetMorganFingerprint, nBits=1024, radius=2)
# calculate fingerprint and get solubility class.
trainX = [fpfunc(m) for m in train_mols]
trainY = [val_dict[m.GetProp('SOL_classification')] for m in train_mols]
testX = [fpfunc(m) for m in test_mols]
testY = [val_dict[m.GetProp('SOL_classification')] for m in test_mols]
rfc = RandomForestClassifier(random_state=794)
rfc.fit(trainX, trainY)
RandomForestClassifier(random_state=794)In a Jupyter environment, please rerun this cell to show the HTML representation or trust the notebook.
On GitHub, the HTML representation is unable to render, please try loading this page with nbviewer.org.
RandomForestClassifier(random_state=794)
# https://www.rdkit.org/docs/Cookbook.html
# to get probability of High solubility
def getProba(fp, predctionFunction):
return predctionFunction((fp,))[0][2]
def drawmol(mols, idx):
d = Draw.MolDraw2DCairo(1,1)
_, maxWeight = SimilarityMaps.GetSimilarityMapForModel(mols[idx],fpfunc,
lambda x: getProba(x, rfc.predict_proba),
colorMap='coolwarm',
size=(200,200),
step=0.001,
alpha=0.2)
d.FinishDrawing()
show_png(d.GetDrawingText())
print(mols[idx].GetProp('SOL_classification'))
# Lets view
high_test_mols = [mol for mol in test_mols if mol.GetProp('SOL_classification') == '(C) high']
low_test_mols = [mol for mol in test_mols if mol.GetProp('SOL_classification') == '(A) low']
drawmol(high_test_mols, 1)
(C) high
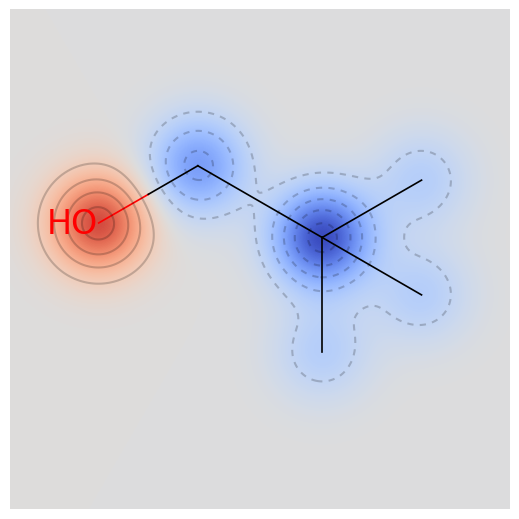
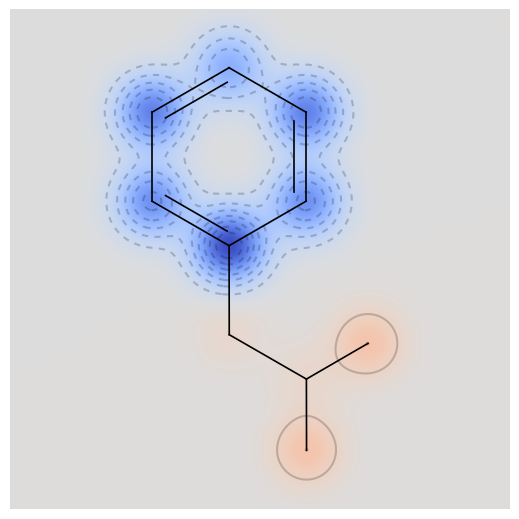
drawmol(low_test_mols, 5)
(A) low
Reference:
http://rdkit.blogspot.com/2020/01/similarity-maps-with-new-drawing-code.html